Were modern humans the first hominids capable of complex spoken language? If so, how did this unique capability evolve? New research by Robert B. Darnell and Erich D. Jarvis helps answer both of those questions, and could further our understanding of language and developmental disorders.
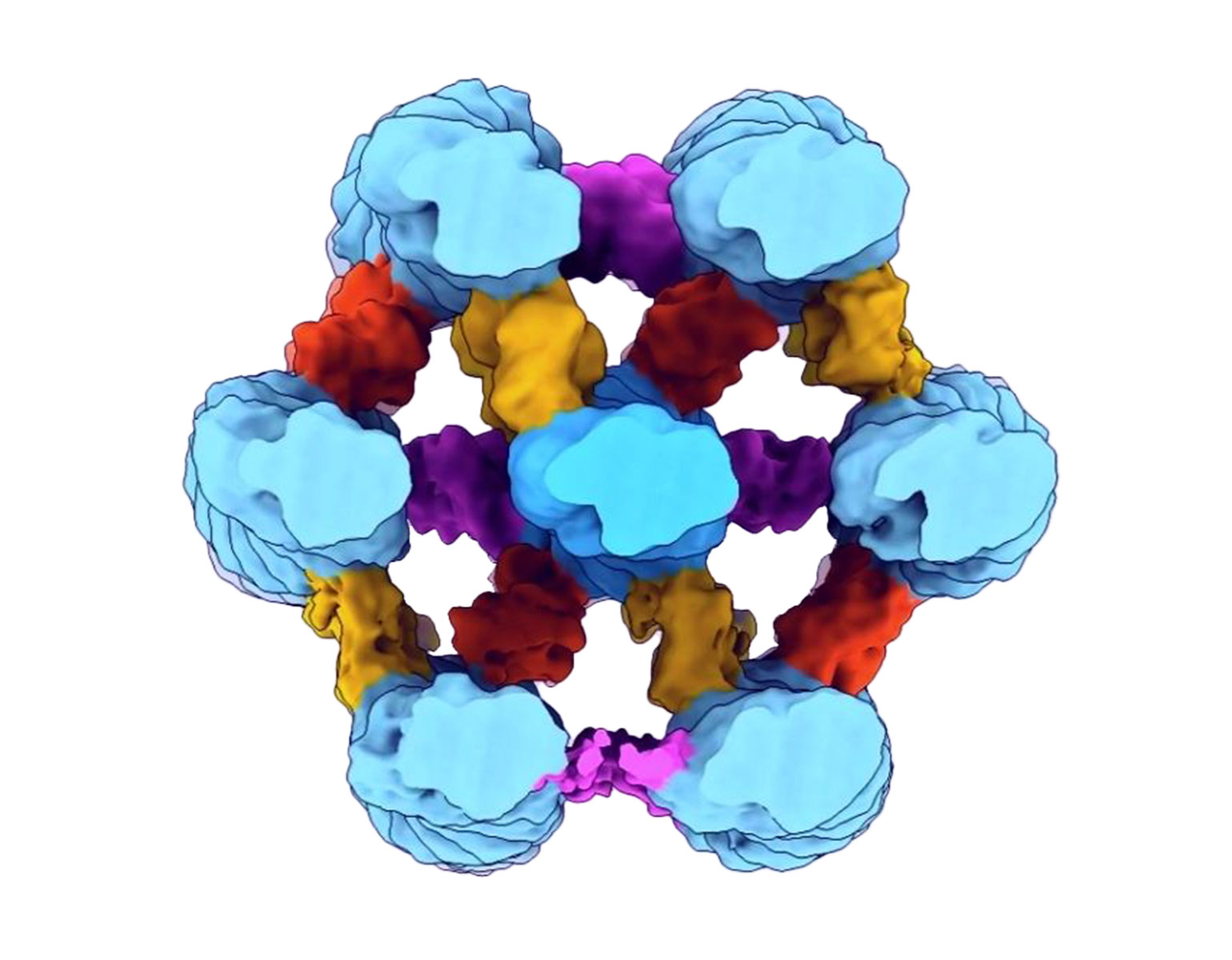
Darnell, who specializes in studying how RNA-binding proteins regulate gene expression, has spent more than three decades investigating a particular RNA-binding protein called NOVA1. NOVA1 is vital to brain development and neuromuscular control—Darnell has identified cases in which variations in NOVA1 are associated with developmental language and motor difficulties—and while it is found in animals ranging from mammals to birds, a particular variant of the protein, known as I197V, appears only in humans.
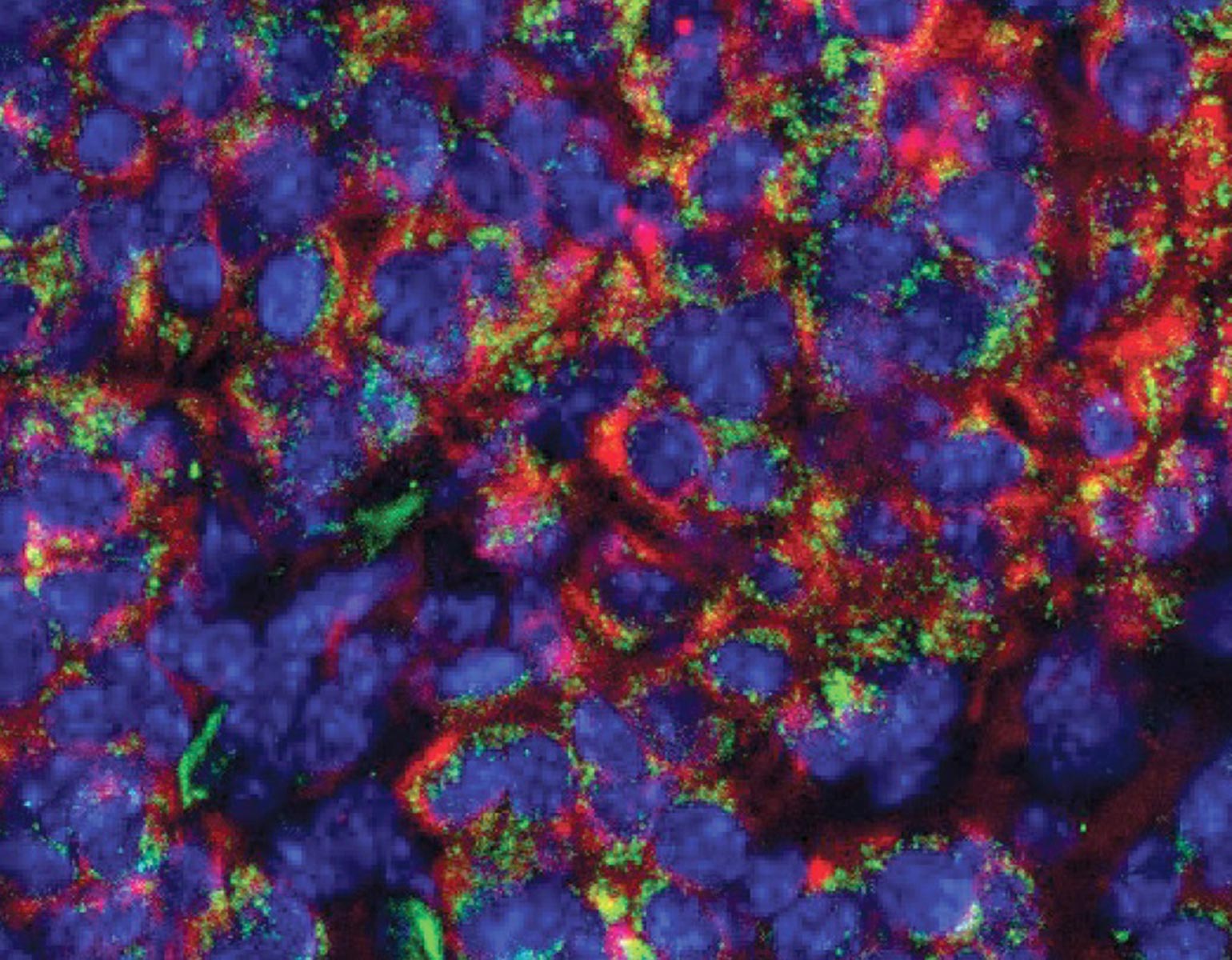
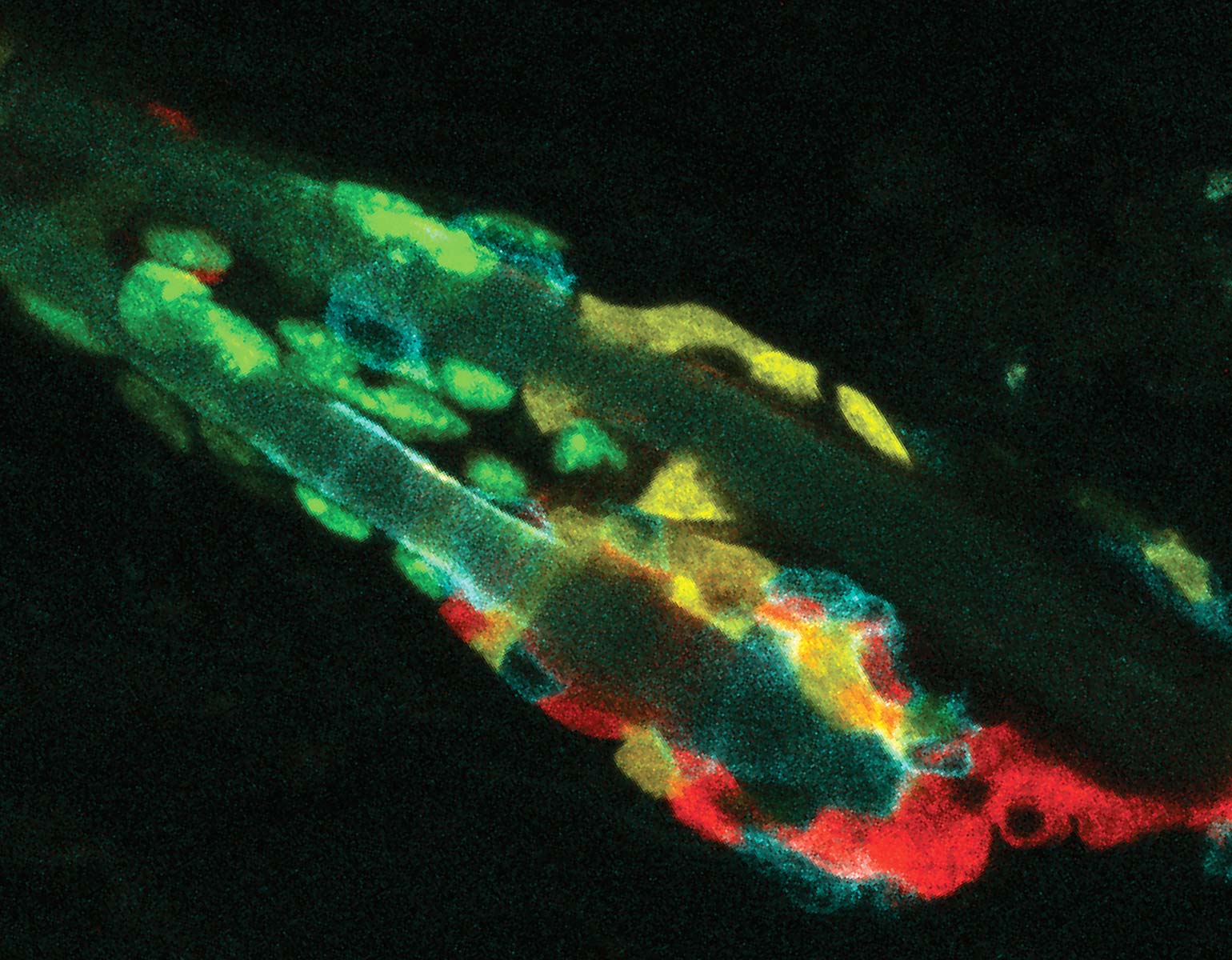
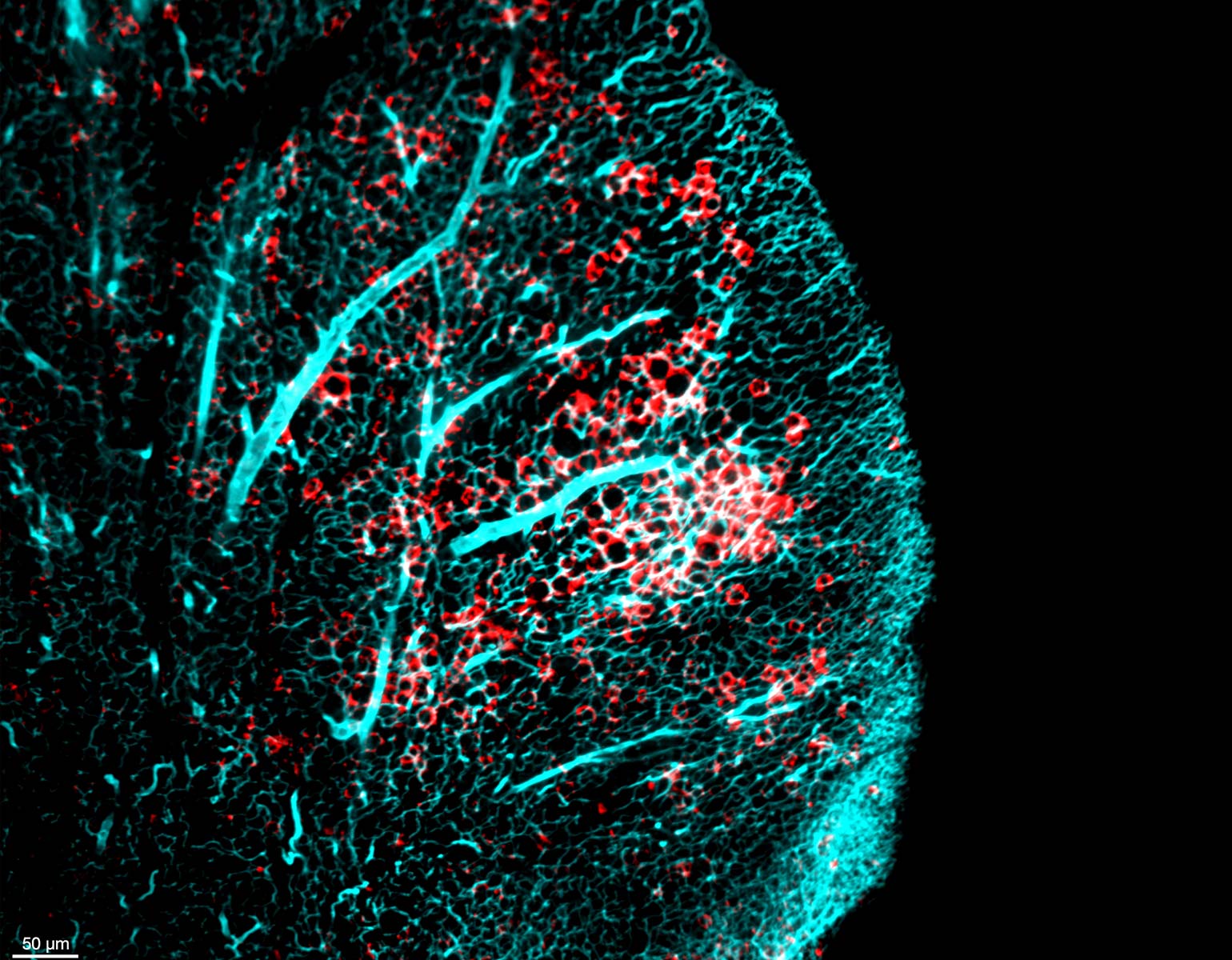
Yoko Tajima, a postdoc in Darnell’s lab, used CRISPR gene editing to replace the common NOVA1 protein found in mice with I197V. Intriguingly, the human-specific variant specifically affected RNA binding at sites related to vocalization.
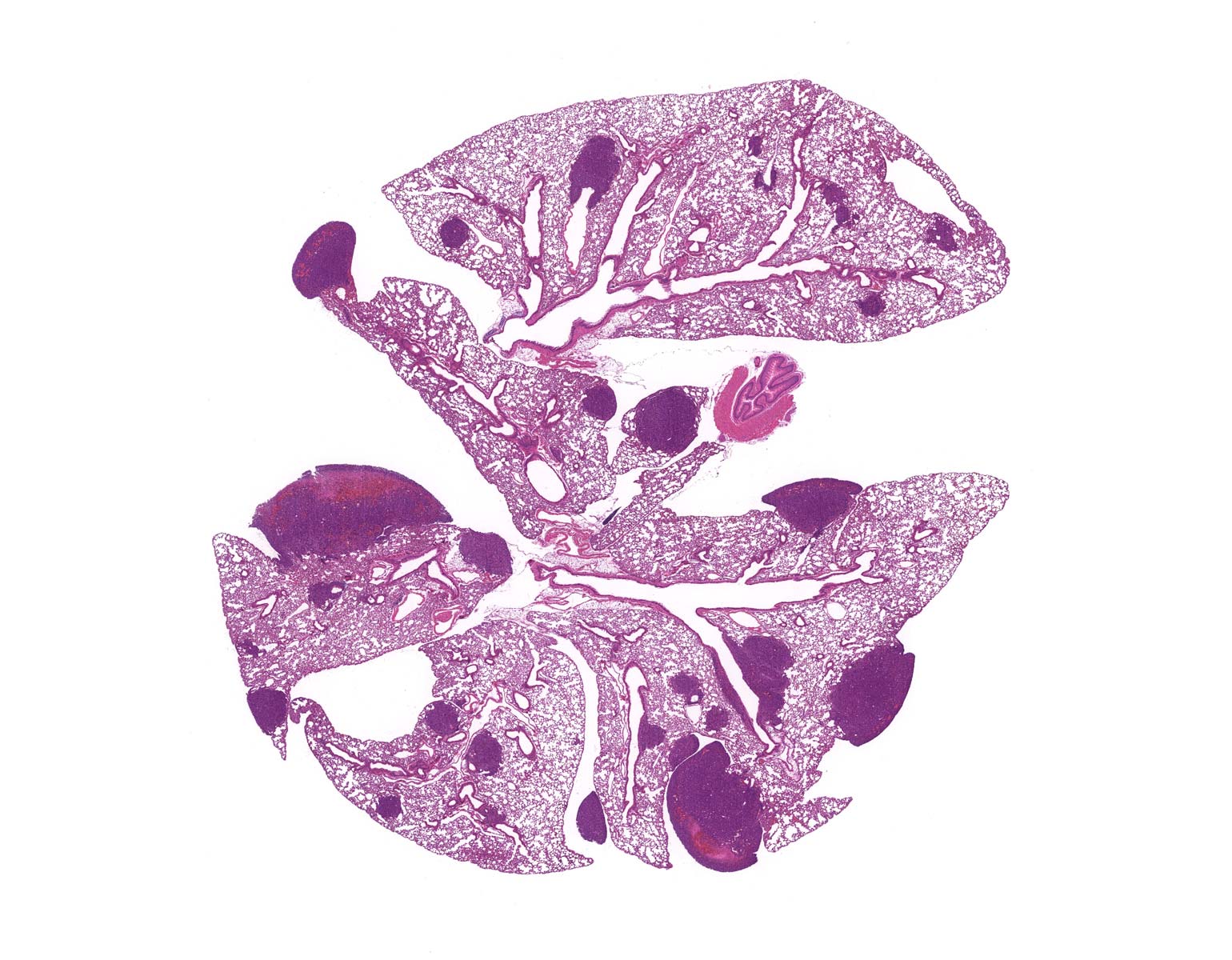
Probing deeper, Darnell joined forces with Jarvis, who studies the molecular and genetic mechanisms underlying vocal learning. Over the next few years, the researchers documented altered vocal patterns among adult male mice and mouse pups of both sexes that carried the human variant.
“The single amino acid change in NOVA1 may make it a bona fide human ‘language gene,’” Darnell says. “Though certainly it’s only one of many human-specific genetic changes.”
To understand the potential influence of I197V on human evolution, the team compared the genomes of modern humans with those of our nearest relatives, the hominids known as Neanderthals and Denisovans. While these archaic relatives had the same version of NOVA1 found in nonhuman animals, the human-specific I197V variant was found in 650,052 of 650,058 modern human genomes analyzed, underscoring how it has become nearly ubiquitous, and suggesting it arose early in Homo sapiens’ evolution.
“Our data show that an ancestral population of modern humans in Africa evolved the human variant I197V, which then became dominant perhaps because it conferred advantages related to vocal communication,” Darnell says. “This population then left Africa and spread across the world.”
The team’s findings advance our understanding of when and how humans acquired their unique linguistic abilities. And by clarifying the role that NOVA1 plays in regulating language along with neural development and motor control, Darnell and Jarvis could also help scientists better understand a wide array of illnesses and impairments.
“Our discovery could have clinical relevance, ranging from children with language and developmental disorders to neurodegenerative disease,” Darnell says.