
Feature
A new
approach
to Alzheimer’s
is unfolding
First, it was all about the plaques. Then plaques, and maybe tangles. Now, a plethora of new ideas are galvanizing efforts to save neurons from decay.
By Joshua KrischThe first new Alzheimer’s drug in 17 years should have been a blockbuster. But when the Food and Drug Administration approved aducanumab in the summer of 2021, it instead made headlines as yet another letdown in a decades-long quest for treatments against the disease. Because while clinical data showed that the drug did precisely what it was supposed to do—remove protein clumps from sick brains—it also failed where it counts most.
Patients were not getting any better.
“It was very disappointing,” says Hermann Steller, Strang Professor at The Rockefeller University. “The field of Alzheimer’s research has been focused on protein aggregates for decades; $30 billion has been poured into it. And there is very little to show for it.”
Aducanumab was developed based on a hypothesis that has steered Alzheimer’s research for decades. Alluring in its simplicity, this idea holds that neurons die mainly because the brain gets clogged by protein waste called beta-amyloid plaque and that clearing that waste, or preventing it from forming in the first place, will cure the disease.

But aducanumab was far from the first drug of its class to fail. Until recently, no treatment designed to rid patients’ brains of plaques had turned out to meaningfully stop decline. And while the latest option on the horizon—a newer amyloid-clearing antibody called lecanemab—has shown some potential in late-stage trials, it also raises serious safety concerns. Even as it was unveiled, many scientists expressed only the most cautious optimism that treatments targeting plaques alone will be the answer to one of the most dreaded diseases on the planet. And for now, roughly 50 million patients around the globe continue to suffer, degenerating through memory loss, dramatic personality changes, and hallucinations, until those who live long enough to reach end-stage disease finally lose the ability to even speak or move.
Why has progress been so excruciatingly slow in the field of Alzheimer’s, even after decades of scientific struggle?
For one thing, brain disorders in general—and neurodegenerative disorders like Alzheimer’s and Huntington’s in particular—present some of the most vexing challenges in all of medicine. By the time Alzheimer’s symptoms manifest, for example, the brain is often too far gone to treat, and events driving the earliest stages of neurodegeneration have lain beyond scrutiny. Access alone presents a major obstacle: The strong bony helmet that protects our gray matter from trauma also makes it impossible to analyze brain tissue, and the barely permeable blood-brain barrier all but seals the brain off from the effects of would-be therapeutics.

And the scientific challenges continue. “Mouse models that are so critical for research on other conditions often fail to capture the nuances of human neurocognition and how it deteriorates due to aging or disease,” says Nathaniel Heintz, Rockefeller’s James and Marilyn Simons Professor and director of the university’s Zachary and Elizabeth M. Fisher Center for Research on Alzheimer’s Disease. “What’s left to look at is postmortem human brain tissue, which historically couldn’t tell us much about what sparked neurodegeneration in the first place.”
Yet there are growing reasons to believe such historical challenges may become a thing of the past. Heintz and Steller, along with other scientists from various fields, are finding new ways to think about the disease, challenging long-held assumptions about amyloid and the very nature of neurodegeneration. This budding movement has all but discarded the notion of Alzheimer’s stemming from a singular origin and replaced a once-dominating approach with a string of others. By focusing on how the brain interacts with multiple systems throughout the body and how the most overt organ decline may arise from the subtlest molecular disruption, these researchers are discovering multiple new avenues for treatment and developing new tools poised to take the study of neurodegeneration to the next level.
“When research on human neurodegeneration started decades ago, all they were able to see was the accumulation of plaques and tangles because that’s what was visible using the technology of the day. So targeting beta-amyloid and tau made sense,” says Heintz, referring to the main ingredients of protein clumps found in brains ravaged by Alzheimer’s. “That was the most precise information available, and the amyloid hypothesis was generated from that data.”
Trained as a molecular biologist, Heintz became interested in neurodegeneration about 10 years ago, bringing a new perspective to the study of brain disease. “Neurons in the brain function for decades and then suddenly succumb,” he says. “Well, that looks to me like a biochemical problem in the cell.” He recognized that without carefully examining that problem at the cellular and genetic level, scientists would keep stumbling in the dark in their efforts to prevent the cells from dying off.
Scientists are focusing on how the most overt organ decline may arise from the subtlest molecular disruption.
But to even begin hunting for the root causes of neuronal decline, Heintz would need two things in exceedingly short supply: human data and, crucially, the ability to tell brain cells apart from one another. There are between 500 and 1,000 cell types in the brain, each with different molecular properties and gene expression patterns, making it extremely difficult to pinpoint which cells are malfunctioning in mixed samples. To sort out the good cells from the bad, Heintz teamed up with the late Nobel laureate Paul Greengard to develop a transformational methodology they dubbed translating ribosome affinity purification (TRAP) because of how it homes in on a cell’s protein factory, the ribosome.
TRAP judges a cell’s character by its actions, clocking every protein it produces as a proxy for its type. Heintz’s group could now not only differentiate between the actors but also use their molecular signatures to identify who was disrupting the show—whether this immune responder, not that neuron, flubbed its lines—delivering an extraordinary tool for his own lab and others in the field that had been spinning their wheels. The technique has already yielded tangible benefits. Heintz and Greengard, along with their Rockefeller colleague Jeffrey M. Friedman, initially used their new method to develop a cell-specific therapy addressing motor-related symptoms in Parkinson’s disease and have since expanded on that work to study addiction, anxiety, and depression.

But Heintz sensed that Huntington’s and Alzheimer’s would be tougher nuts to crack. Because TRAP works best in animal models, his team went on to develop another tongue twister, fluorescent-activated nuclei sort sequencing (FANSseq). It works off a similar principle but with more subtlety, allowing scientists to characterize a cell by tracking the genes expressed in its nucleus. Only after applying FANSseq to the study of Huntington’s—a comparatively straightforward disease caused by a known gene—did Heintz feel ready to set his sights on the thorny ambiguities of Alzheimer’s.
Of all the unknowns surrounding this disease, the most confounding is how and when it starts. Scientists know it takes decades to develop, but no one has been able to pinpoint the precise genesis. Charting molecular changes over time would allow researchers to piece together the sequence of events that ultimately leads to full-blown disease, and this is where Heintz hoped his years of tracking biochemical nuance would give him an edge.
One way of looking for these early stages is to examine brain tissue donated by patients with Alzheimer’s who die of other causes, like accidents or heart attacks, and then compare it with that of donors without Alzheimer’s.
Five fragile neurons
There are hundreds of neuronal cell types in the brain and hundreds of diseases in which one or several of these cell types fail—leading to problems with memory, mood, movement, and more. Here are some of the types most famously prone to decay.
Illustrations by Kasia Bogdanska
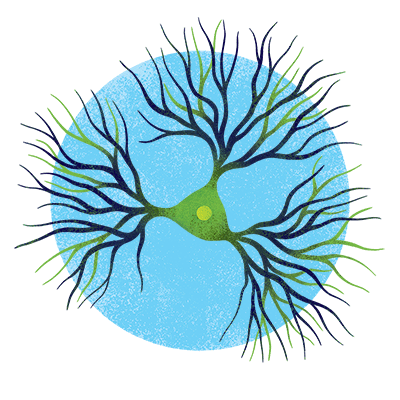
Pyramidal neurons
Named for their shape, pyramidal neurons kick-start numerous cognitive and motor functions. Alzheimer’s pathology disproportionately impacts these cells for reasons yet unknown.
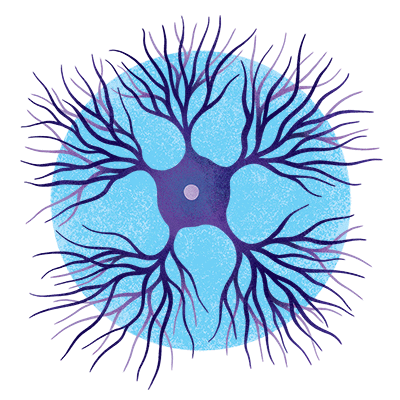
Medium spiny neurons
These cells live mostly in the striatum, the brain’s headquarters for motor and reward systems, and act as a circuit breaker to quiet neural activity as needed. They are the first cells to die in Huntington’s disease, a condition caused by an inherited mutation.
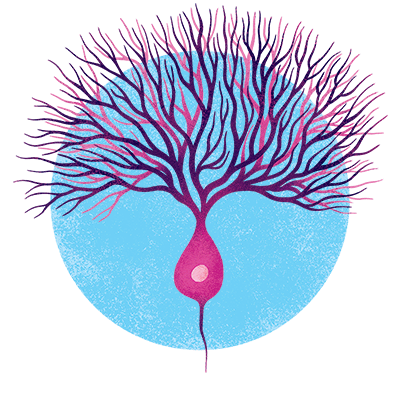
Purkinje neurons
These beautiful, bushy cells are found in the brain’s cerebellum—a region responsible for movement, cognition, and emotion—as well as in the spinal cord. They deteriorate in a group of inherited neurodegenerative diseases, called spinocerebellar ataxia, leading to problems with movement coordination, among other things.
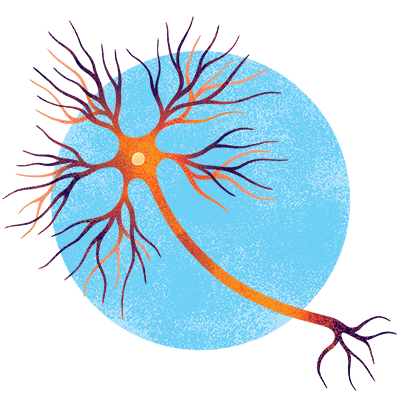
Motor neurons
These cells carry out voluntary movements like walking and talking, as well as involuntary ones like breathing. Their long neural extensions connect the brain and spinal cord with muscles throughout the body, some reaching all the way to the toes. When motor neurons degenerate in amyotrophic lateral sclerosis (ALS), muscles gradually weaken and ultimately atrophy.
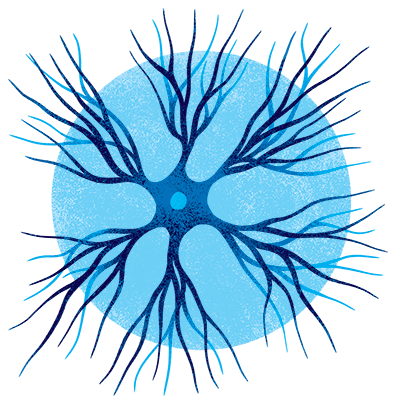
Dopaminergic neurons
Named for their ability to produce dopamine, a neurotransmitter playing a decisive role in mood, stress, and addiction, these neurons also coordinate voluntary locomotion. Their loss contributes to the distinctive movement disorders of Parkinson’s.
By using FANSseq to examine post-mortem tissue, the Heintz lab can study the state of each brain cell as it progresses through these diseases, generating findings that are helping researchers paint a fuller picture of how things begin to go wrong. In particular, studying why certain cell types are consistently the first to die off could point the way to slowing or stopping degeneration. There, Heintz has already spotted parallels with Huntington’s, noting that the neurons that perish earliest are located in the cerebral cortex of Alzheimer’s patients, right beside those most vulnerable in people with Huntington’s. “It’s a beautiful demonstration of how specific this pathophysiology is,” says Heintz. “Our approach could soon provide additional insight into known mechanisms and reveal features that could offer new options for therapy.”

At least one emerging pathway for treatment may come from a new twist on an old idea: While amyloid specifically may not prove to be the key cause of Alzheimer’s disease, proteins in general could in fact be a culprit.
A cell’s proteins live lives of constant flux—assembling, activating, and continuously adjusting until finally wearing out. At that point, each must be swiftly dismantled. If that process goes awry for any reason, toxic protein waste may accumulate throughout the body, potentially leading to muscle-wasting disorders or neurodegeneration.

Steller was decades into his research on how the body breaks down excess proteins and why those systems sometimes fail when he was struck by the larger implications of a serendipitous discovery.
“It was an opportunity to make a contribution to the field of age-related neurodegeneration and offer a completely new explanation for how and why these diseases start,” he says.
Steller had been studying proteasomes, the molecular machines that degrade proteins, when he discovered how these disassemblers travel long distances inside neurons. Proteasomes are made in the neuron’s main body, but must do their jobs at the tips of its extensions, the long dangling threads that send and receive signals. If the neuron were the size of a basketball, Steller posits, its extensions would reach halfway across the width of Manhattan.
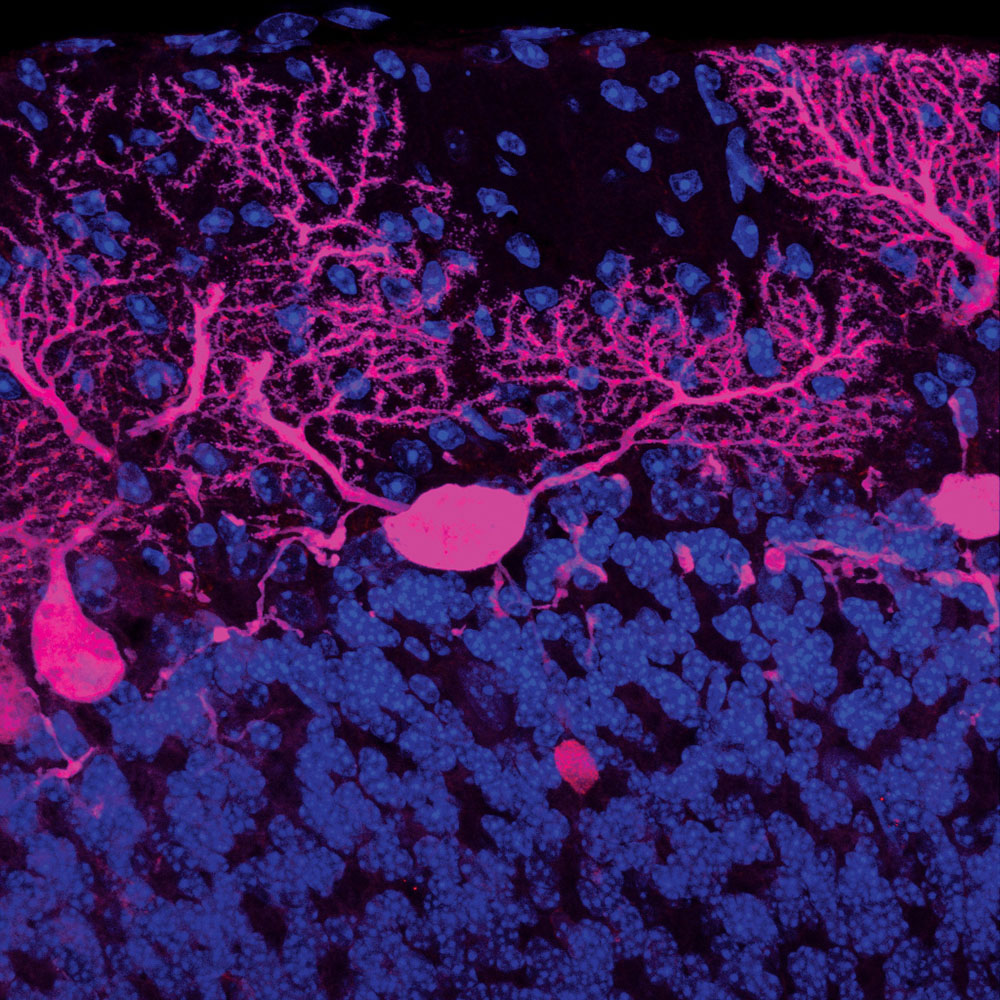 Damaged cerebellum
Damaged cerebellum
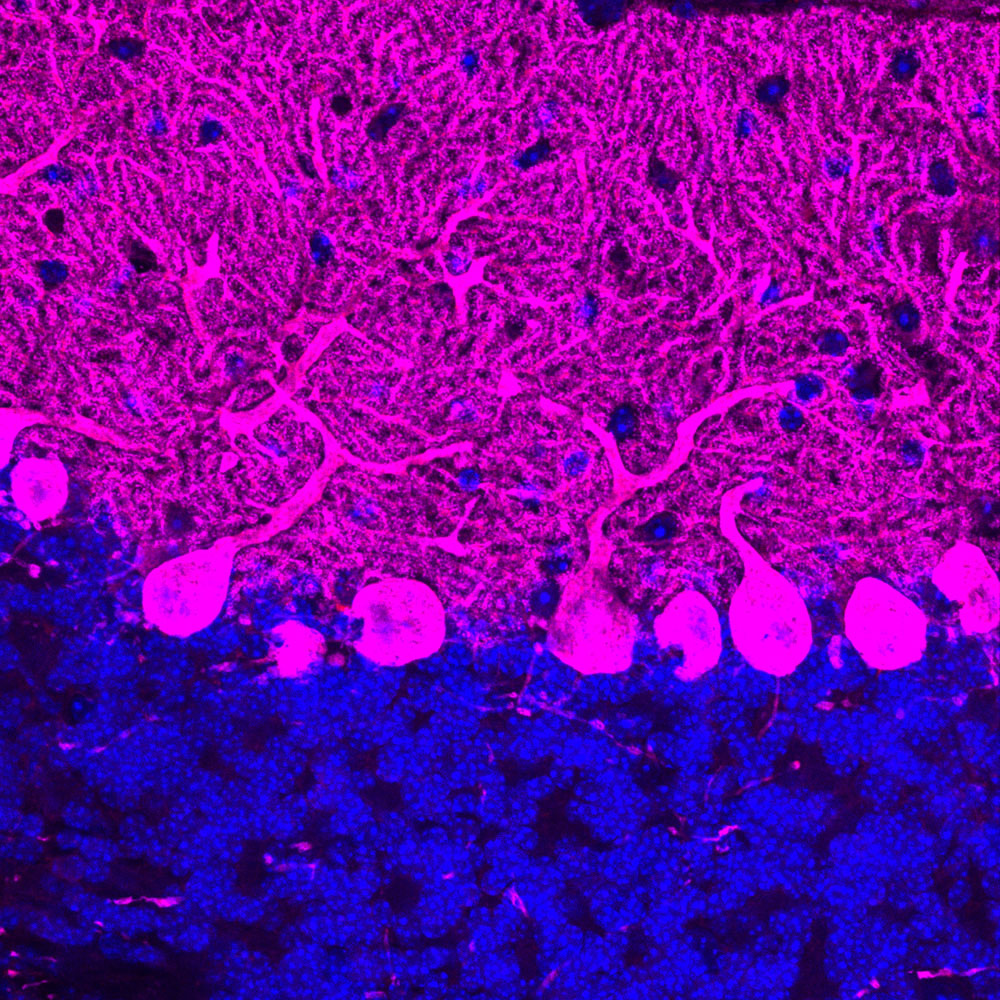 Healthy cerebellum
Healthy cerebellumTo get to where they need to be, proteasomes get loaded onto transporters—“like children on a school bus,” Steller says—that ferry them across town. Recently, Steller’s team reported that as the brain ages, the proteasome transporters become unable to keep up with increased demands for protein degradation. Over time, that translates into a progressive increase in protein damage as more stress gets added to the waste removal system. Eventually, there’s a tipping point when there are no longer enough garbage collectors available to handle all the detritus piling up at the nerve endings. “We think that Alzheimer’s, Parkinson’s, ALS, and many other aging-related diseases may boil down to this system not working well enough as we age,” Steller says. “It really is a weak link.” When proteasomes fail to reach their destination, these accumulations can prevent neurons from communicating with one another, ultimately causing them to die.
“Many aging-related diseases may boil down to the protein removal system not working well enough as we age.”
There is ample support for Steller’s hypothesis, including that people with a mutation known to stymie the activity of a proteasome regulator known as PI31 are predisposed to Parkinson’s disease and that mutations in PI31 itself have been linked to Alzheimer’s. And when the researchers jump-started the proteasome transport system in a mouse model of Parkinson’s, this prevented neurodegeneration and greatly delayed onset of the disease.
Steller’s lab recently devised a strategy to bolster that same pathway in human cells, with the ultimate goal to develop novel therapies for the treatment of neurodegenerative diseases. “We believe we’ve found a root cause of why we see declining function in neurons as we age,” Steller says—a finding that has implications not only for neurodegenerative diseases but for the aging brain overall.

“Isn’t it stunning?” asks research associate professor Erin Norris, pointing to a bright crimson image depicting the mazy thicket of the cranial vascular system, which hangs on the wall of the Patricia and John Rosenwald Laboratory of Neurobiology and Genetics. Indeed, even with all the gray matter removed, it is easy to recognize the shape of a human brain just by the outline of the countless vessels that feed it. No other organ is so inextricably intertwined with our vascular system, nourished as it is by more than 400 miles of capillaries. And this makes the brain particularly vulnerable to vascular disorders, including those involving inflammation.
A first glimpse of the disease
Alois Alzheimer first laid eyes on a diseased brain in 1906, shortly after the death of one of his patients. Auguste Deter had been a poor homemaker suffering from memory problems, paranoia, and aggression. Alzheimer, a young psychiatrist with a burning interest in anatomy, placed a section of Deter’s brain under his microscope. Gazing into the lens, he saw tissue clogged with thick deposits that would later be identified as amyloid plaques, and neurons notched with telltale whorls now known as neurofibrillary tangles.
Alzheimer believed he’d pinpointed the pathology behind a form of dementia, what he called “an unusual disease of the cerebral cortex.” A few years later, the condition made its formal debut in a psychiatry textbook authored by his mentor, Germany’s then-leading psychiatrist, who named it after Alzheimer.
Throughout the next century, researchers would chase the plaques and tangles that Alzheimer had painstakingly drawn in his case notes. In the 1980s, scientists identified the plaques as aggregates of a peptide known as beta-amyloid and the tangles as dysfunctional mutations of the protein tau.
A highly choreographed process, inflammation is how the immune system cordons off an injury, rushes blood and other fluids to the site, and bombards it with scavengers seeking to destroy the threat, be it pathological (a nasty germ) or physical (a painful splinter). Once the invader has been neutralized, the process shuts down. Sometimes, however, it gets triggered not by an external force but by an internal misfire, as with chronic inflammatory disorders such as lupus and Crohn’s disease. Instead of healing the affected tissues, inflammation can then run amok, wreaking damage. Wherever it manifests, leaky vessels and dysfunctional clotting ensue.
Norris and her colleagues have long focused on clotting and inflammation, paying particular attention to Alzheimer’s once they began to see striking findings emerge from a series of experiments designed to explore how coagulation and inflammation intersect with the disease.

Unexpectedly, this work revealed that knocking down a key component in the inflammation system rendered cells in the brain’s hippocampal region resistant to decay. It was a pivotal moment. “We knew early on that the vascular system was involved in neurodegeneration,” says Sidney Strickland, the Zachary and Elizabeth M. Fisher Professor in Alzheimer’s and Neurodegenerative Disease, who heads the lab. “We just didn’t know how.”
The scientists began looking into familial Alzheimer’s, a rare genetic version of the disease that strikes early and, tragically, is inescapable for those carrying the gene. “There are some people who we know, at birth, will invariably get Alzheimer’s at around age 50, and there’s no escape,” Strickland says. Such patients offer scientists a unique window into the earliest stages of neurodegeneration—what the brain and body of a future Alzheimer’s patient look like in childhood, adolescence, and early adulthood.
“Everything doesn’t just collapse at age 50 with these patients,” Strickland says. “It’s been shown that things look abnormal decades before patients start experiencing cognitive impairment.”
One glaring abnormality: high levels of deposited fibrin, the major protein component of blood clots in the brain. “We soon discovered it wasn’t just higher levels of fibrinogen itself in early Alzheimer’s,” Norris says, “but also changes in inflammation, coagulation, and blood flow to the brain that seemed to come along with it.”
“We aren’t saying that amyloid plaques aren’t involved in Alzheimer’s,” Strickland says. “We’re saying that targeting plaques is not the only approach for treating this disease.”
“Many aging-related diseases may boil down to the protein removal system not working well enough as we age.”
It makes sense that inflammation would promote neurodegeneration. “Neurons are fragile, delicate cells,” says Marc Flajolet, a research associate professor in the lab formerly led by Greengard. With their exceptionally high energy requirements, neurons are frighteningly easy to kill and “are the first to die if there’s a chronic problem,” Flajolet says. One theory is that molecules secreted during chronic inflammation might be toxic to neurons; another line of reasoning goes that inflammation recruits molecules that eat up synapses, disconnecting neurons from one another and causing them to die.
Although these ideas were initially met with some resistance, researchers now generally embrace the importance of abnormal clotting and inflammation in Alzheimer’s disease. Part of that acceptance came on the heels of another advance by Strickland’s lab—the creation of a therapeutic antibody that mutes the plasma clotting system. When the scientists tested this drug in mice, they found that it significantly reduced the animals’ inflammation and coagulation profiles. The team is now advancing their animal trials of the antibody with an eye toward moving it into the clinic.
It remains to be seen whether the classic drug-development approach inspired by the amyloid hypothesis will on its own make a meaningful difference for patients. Whatever the case, important questions remain surrounding amyloid and its fellow traveler, tau. Flajolet’s work, for instance, is predicated on the assumption that protein aggregates actually are the main problem—just not all aggregates of all sizes. “We believe tau protein aggregation, specifically, may be central to where and when Alzheimer’s disease starts and to how it evolves and progresses over time,” he says.
Until recently, tau played second fiddle to amyloid at least partly because there was no easy way to study its deposits in the brain, known as tau tangles, which are quietly ensconced within damaged brain cells (amyloid plaques, by contrast, tend to overtly gum up the cells’ exterior). But then the FDA approved a new intravenous drug in 2020 that allowed physicians to track tau aggregates in a patient’s brain and to image tau pathology via PET scan. Researchers now believe that the largest deposits—the infamous neurofibrillary tangles—are not where most of the danger lies.

“We believe the toxicity is not so much coming from fully formed neurofibrillary tangles as from their precursors, the smaller oligomers,” Flajolet says. His team is currently looking for molecules that might block the formation of small tau oligomers, slow their aggregation, interfere with their spreading, or at least make them easier to track and study in the future.
Truth be told, even those scientists shifting away from amyloid and tau remain interested in the storied plaques that kicked off the study of neurodegeneration and all but defined the field until recently. “What we’re finding isn’t running counter to conventional wisdom so much as adding nuance and value and insight,” Heintz says.
The widening scope of Alzheimer’s research has yet to bear fruit in the form of novel therapies. But with new perspectives and tools in hand, we are in a better position than ever to get to the roots of a problem that has claimed memories, personalities, and lives for millennia—and, ultimately, to understand how that problem might be fixed.
“Maybe it’ll be neuroinflammation or a new take on tau or something we haven’t even considered yet,” Heintz says. “But I have no doubt we’re going to see new and effective therapies unfold in the decade ahead.”