Feature
Against the grain
For years, people thought Sidney Strickland was barking up the wrong tree. He wasn’t.
By David NoonanSidney Strickland knew what he was up against when he began working on the disease more than two decades ago, and little has changed: Alzheimer’s remains a deadly health threat and among the most feared diseases on the planet, unchecked in its power to destroy brain cells and erase minds.
In the last 15 years, more than 400 new Alzheimer’s drugs have failed clinical testing in humans. That’s 400 times that the hopes of scientists and doctors, along with those of patients and their families, have been squashed.
But if the harsh reality of Alzheimer’s hasn’t changed, something else has. Scientists are finding new ways of thinking about the disease and studying its biology, thanks in part to a question that Strickland began asking in the 1990s.
What role, Strickland wanted to know, do impairments in the brain’s blood supply play in Alzheimer’s? At the time, Strickland, now a Rockefeller scientist and head of the Patricia and John Rosenwald Laboratory of Neurobiology and Genetics, was working on problems related to the circulatory system at the State University of New York at Stony Brook.
It was a new idea, a question that had not been asked before. But over the past two decades, Strickland’s ongoing efforts to answer it have opened up whole new avenues of research into the nature and causes of Alzheimer’s, which currently affects 5.7 million Americans, and is expected to reach nearly 14 million by 2050.
Unlike most people in the field, Strickland has no formal training in brain disease or neuroscience. He is a developmental biologist who has climbed a steep learning curve, motivated in part by his concern that a single disease pathway had come to dominate an entire field.
“I think Alzheimer’s has suffered from oversimplification,” he says.
To this day, most Alzheimer’s research focuses on a sticky protein called amyloid-beta, the accumulation of which leads to the formation of gummy plaques in the brain. To be sure, amyloid-beta deserves the attention: It has been shown to drive the development and progression of Alzheimer’s and scientists have found that the plaques can interfere with neurons’ ability to send signals, as well as sentence them to an early death. The question is whether other types of brain changes help fuel the disease as well, and Strickland thinks there is much to be gained from thinking more broadly. Like cancer, he says, Alzheimer’s is fundamentally complex and may arise from multiple pathogenic pathways. And one mechanism that has been largely overlooked involves irregularities in the brain’s vascular system.
In particular, Strickland has zeroed in on the brain-damaging effects of fibrinogen, a protein that gives rise to blood clots. Under normal circumstances, fibrinogen, which circulates in the bloodstream at high concentrations, is beneficial: Whenever a blood vessel gets damaged, a cascade of molecular events is triggered to convert it into fibrin, a mesh-like substance that stops bleeding and initiates repair of the vessel wall.
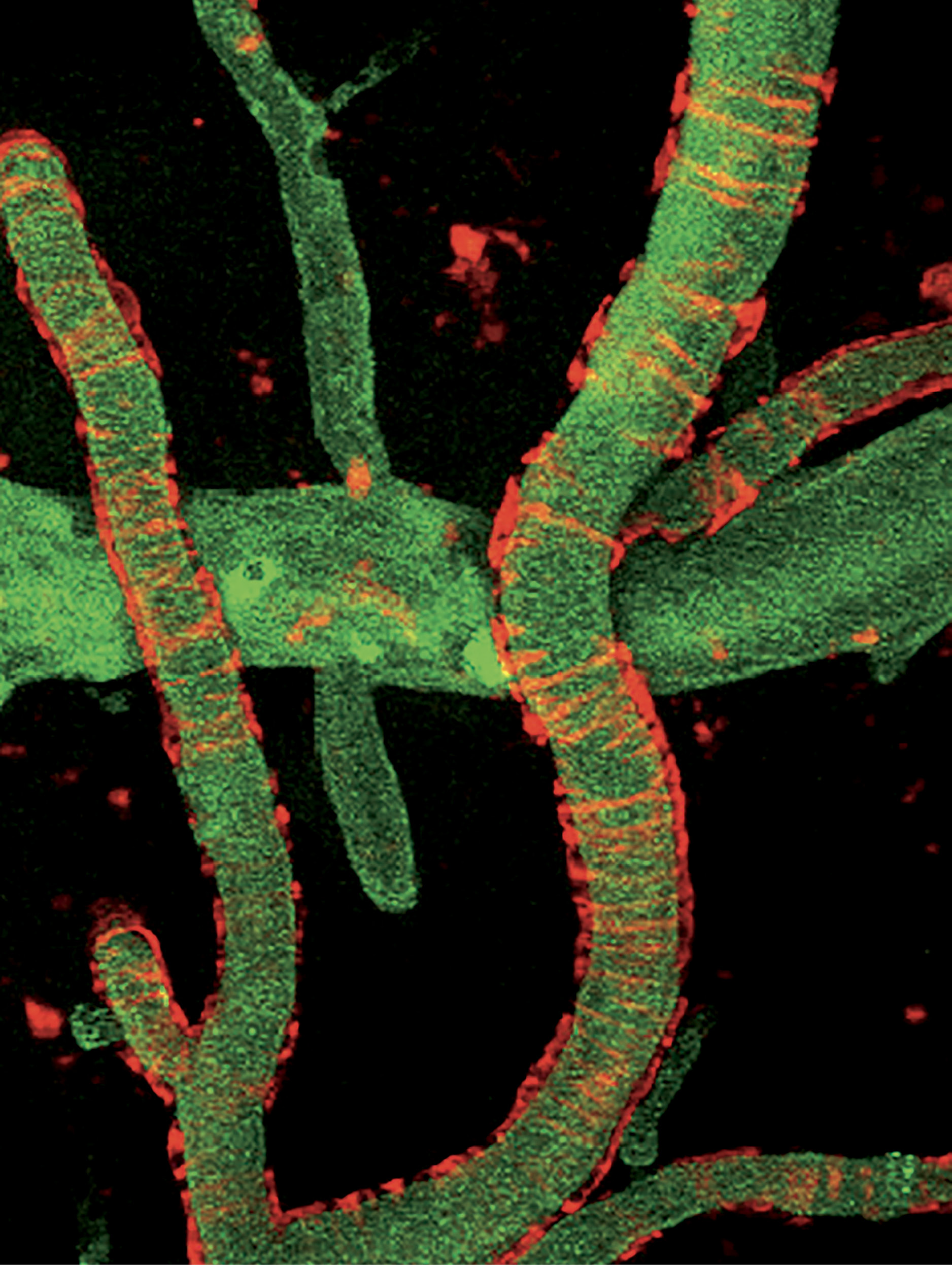
However, as Strickland has discovered, fibrinogen can sometimes leak into the brain, where it does not belong, and cause fibrin to accumulate. In an analysis of postmortem brain tissue, Strickland found fibrin buildups in multiple areas of the brains of people with Alzheimer’s—much more fibrin than would be expected in healthy brains. In the hippocampus, a part of the brain essential for memory, Strickland and his colleagues discovered more than 20 times as much fibrin, and in the superior frontal cortex, which is involved in many higher cortical functions, 100 times as much.

Exactly how fibrinogen seeps into the brain is something of a mystery. To get there, the protein needs to cross the blood-brain barrier, the brain’s primary defense system. “The blood-brain barrier is made up of several different types of cells,” says Erin Norris, research assistant professor in the lab. “Presumably, something goes wrong in some aging brains, and these cellular components of the blood vessel wall start to break apart.” The rupture allows fibrinogen to gain access to the interior of the brain.
It’s unclear at this point exactly when in the course of Alzheimer’s fibrin deposits become a factor and whether they are a primary cause of the disease or a consequence of other, earlier pathogenic mechanisms. What they are not, says Strickland, is a mere comorbidity, a separate condition that happens to accompany aging. Leaking fibrinogen and fibrin clot formation contribute to Alzheimer’s, he says, by increasing neurovascular damage, neuroinflammation, and neuronal degeneration, as well as contributing to the deposit of amyloid-beta in and around blood vessels. That assertion is supported by a set of observations his lab made in mouse models of the disease—mice genetically engineered to develop Alzheimer’s. In those experiments, they found that fibrin deposits in the brain increased over time and correlated with the level of amyloid-beta plaques. Conversely, decreasing fibrinogen levels in the Alzheimer’s mice reduced neuronal death in the hippocampus.
The mechanisms by which fibrin accelerates neuronal degeneration remain unknown, but the scientists have a few promising leads. Inflammation, commonly found in the brains of Alzheimer’s patients, is a likely contributor, and Strickland points to the interaction between fibrin and amyloid-beta in the brain as a major source of that inflammation. That interaction, which Strickland has analyzed in detail, is what slows the breakdown of blood clots, a process the body normally undertakes after a wound has healed, when the integrity of the blood vessel has been restored and the clot is no longer necessary. But amyloid-beta disrupts this natural system and prevents fibrin aggregates from dissolving normally.
The result is an ever-increasing load of fibrin that may lead to chronic inflammation. While the protein’s pro-inflammatory function is normally beneficial—it is part of the body’s wound-healing process—the chronic inflammation that ensues when fibrin lingers in the brain can lead to cellular damage.
Another way renegade fibrin clots could kill neurons, says Strickland, is by collecting in and around blood vessels, reducing or even blocking the flow of blood to brain cells, a problem known as ischemia. “If the ischemia is happening in micro-vessels, capillaries, rather than in veins or arteries, then you’re not going to collapse from a stroke,” Strickland says. “You’re going to lose one neuron at a time. And over the course of decades, enough neurons die.”
Lingering lattices
Amyloid-beta, the protein responsible for plaques in the brains of people with Alzheimer’s, also affects blood circulation. It binds to fibrin, the substance that forms blood clots, making the the clots harder to eliminate.

In clots that lack amyloid-beta, the fibrin mesh looks tidy and is readily degradable.
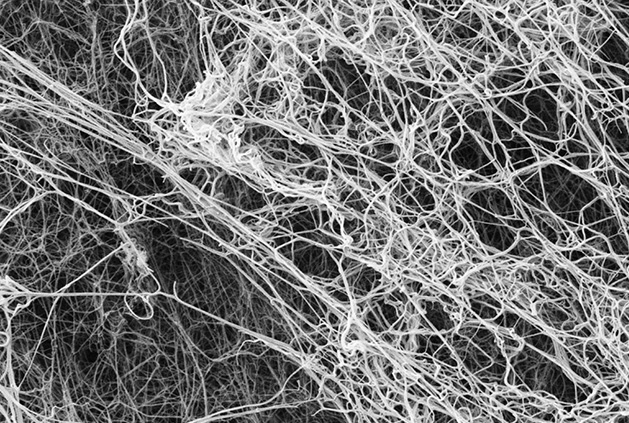
When amyloid-beta is added, the fibrin structures are tangled and don’t easily break down.
Though it left him well outside the mainstream of Alzheimer’s research, Strickland’s decision in the 1990s to investigate the role of cerebrovascular dysfunction in the disease was based on more than mere intuition. As he points out, other types of dementia have long been associated with abnormal blood flow in the brain, often caused by stroke, that deprives neurons of oxygen and nutrients. In addition, half of all Alzheimer’s patients were known to have some kind of impaired cerebral circulation. And multiple studies have shown that physical exercise, which improves cerebrovascular health, can decrease the risk of developing dementia and delay the progression of age-related cognitive decline.
Nevertheless, it took many years for Strickland’s lab to be recognized as an important front in the war on Alzheimer’s. Katerina Akassoglou, now a professor of neurology at the University of California, San Francisco’s Gladstone Institute of Neurological Disease, trained with Strickland as a postdoc and was involved in the lab’s earliest work on fibrinogen. “When I would tell other neuroscientists I was working on fibrinogen,” she recalls, “they would say, ‘Oh, we’re so sorry you’re leaving the neuro field.’”
Howard Fillit, a neuroscientist at the Icahn School of Medicine at Mt. Sinai and executive director of the Alzheimer’s Drug Discovery Foundation, has tracked developments in Alzheimer’s for decades. “When research started back in the eighties,” he says, “it was totally focused on amyloid and similar proteins because those were the only clues we had.” So today, the majority of drugs being developed are focused on those proteins. “There was no research on vascular pathology. But Sid was persistent and he did really good work.”
So it’s something of a new era for Strickland, who recently published what amounts to a summation of his Alzheimer’s work to date in the Journal of Clinical Investigation. “I think the pendulum is swinging,” he says. “About 10 years ago I was describing our ideas to the head of an Alzheimer’s foundation. He said I was barking up the wrong tree. Now he supports our work.”

In Fillit’s view, one of the most important aspects of Strickland’s research is the way it establishes cerebrovascular abnormalities—including common aging disorders such as hypertension and atherosclerosis—as part of the pathology of Alzheimer’s. This way of understanding the disease expands the number of possible therapeutic targets and invigorates the search for new drugs. “Just as treating multiple disease mechanisms in cancer has improved outcomes, a similar evolution of therapy can be envisaged for Alzheimer’s,” Strickland says. He and his team are currently looking at antibodies that could inhibit the interaction of fibrinogen and amyloid-beta in the brain.
“Just as treating multiple disease mechanisms in cancer has improved outcomes, a similar evolution of therapy can be envisaged for Alzheimer’s.”
Strickland’s research could also contribute to new methods for diagnosing Alzheimer’s sooner—such as testing patients experiencing cognitive impairment for vascular abnormalities and inflammation—and new ways to track its progression. Multiple studies already have shown that high levels of fibrinogen in plasma increase the risk of dementia, and the protein was recently established as a biomarker for Alzheimer’s.
The disease has so far managed to resist all efforts to disrupt its lethal course, and Strickland may or may not find success where so many others have failed. Whatever happens, he has already succeeded at broadening the scope of Alzheimer’s research and changing the way we think about this maddeningly complex disease. That’s a breakthrough by any measure.